Hazem A Ghabbour1,2 ![]() ,
Maha M Qabeel3
,
Maha M Qabeel3
For correspondence:- Hazem Ghabbour Email: ghabbourh@yahoo.com Tel:+966146 95853
Received: 3 September 2015 Accepted: 5 January 2016 Published: 28 February 2016
Citation: Ghabbour HA, Qabeel MM. Synthesis, crystal structure, density function theory, molecular docking and antimicrobial studies of 2-(3-(4-phenylpiperazin-1-yl) propyl) isoindoline-1,3-dione. Trop J Pharm Res 2016; 15(2):385-392 doi: 10.4314/tjpr.v15i2.23
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To determine the exact structure and antimicrobial activity of 2-(3-(4-phenylpiperazin-1-yl) propyl) isoindoline-1,3-dione.
Methods: 2-(3- (4-Phenylpiperazin-1-yl) propyl) isoindoline-1,3-dione (C21H23N3O2) was synthesized by the reaction of phthalimide with 1,3-dibromopropane to form 2-(3-bromopropyl) isoindoline-1,3-dione, and was then treated with 1-phenylpiperazine in acetonitrile. The structure of the title compound, 2-(3-(4-phenylpiperazin-1-yl)propyl)isoindoline-1,3-dione, was characterized by proton nuclear magnetic resonance spectroscopy (NMR) and single crystal x-ray diffraction method. The target compound was tested for its antimicrobial activities and computational studies including density function test (DFT) and docking studies were performed.
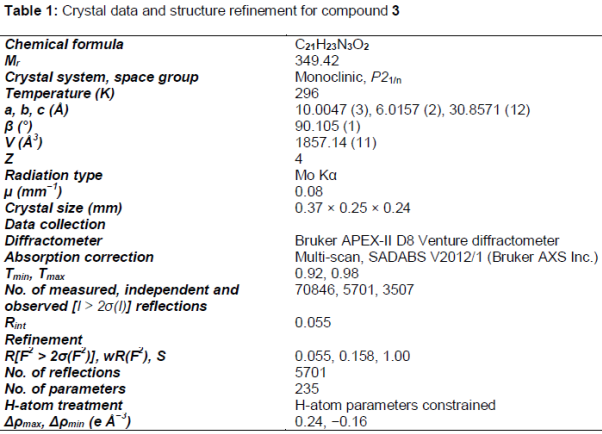
Results: The crystal structure is monoclinic, P21/n, a = 10.0047 (3) Å, b = 6.0157 (2) Å, c = 30.8571 (12) Å, β = 90.105 (1) °, V = 1857.14 (11) Å3, Z = 4, wRref(F2) = 0.158, T = 296 K. The molecules are packed in the crystal structure by non-classical intermolecular C–H….O interactions. Besides HOMO–LUMO energy gap was performed at B3LYP/6-31G (d,p) level of theory. The compound exhibited good activity against S. aureus and C. albicans with zones of inhibition of 15 cm and 18 cm, respectively
Conclusion: The test compound has a moderate antimicrobial activity and the optimized molecular structure of the studied compound using B3LYP/6-31G (d,p) method showed good agreement with the reported x-ray structure.
Introduction
Phthalimide (1,3 isoindolinedione) derivatives have been broadly utilized in medicinal chemistry and drug design because of extensive variety of biological activities such as antimicrobial, anti-convulsant, anticancer, anti-inflammatory, analgesic, hypolipidimic and immunomodulatory activities [1]. The use of phthalimide as primary amine protecting group is extensively reported in the chemical literature, especially in alpha amino acid [2]. On the other hand, N-phenylpiperazine subunit represents one of the most varied scaffolds used in the medicinal chemistry fields [3]. The hybridization between the two groups makes very good chance for many biological activities like human adenosine A2A receptor agonists [4], selective dopamine D3 and D4 agonists [5], selective 5-HT2A receptor antagonists [6] and calcium channel blockers [7].
Methods
Materials and instrument
All organic solvents and used were obtained from commercial sources and used with further purification. Melting points (ºC) were measured in open glass capillaries using Branstead 9001 electrothermal melting point apparatus. 1H (500 MHz) NMR spectra were obtained on Bruker AC 500 Ultra Shield spectrometer (Fällanden, Switzerland). (ESI-MS) was determined using Agilent Ion Trap Mass Spectrometer. X-ray data collection was carried out on Bruker SMART APEX II CCD diffractometer, cell refinement: SAINT [8]; data reduction: SAINT; program used to solve structure: SHELXS; program used to refine structure: SHELXL; molecular graphics: SHELXTL [9]; software used to prepare material for publication: SHELXTL, PLATON [10] and Mercury [11].
Synthesis of the compounds
The 2-(3-(4-phenylpiperazin-1-yl) propyl) isoindoline-1,3-dione was synthesized by the reaction of phthalimide with 1,3-dibromoporpane to give 2-(3-bromopropyl)isoindoline-1,3-dione which in turn reacted with N-phenylpiperzine and potassium carbonate in acetonitrile (Scheme 1).
2-(3-Bromopropyl) isoindoline-1,3-dione (2): 3.20 g (22 mmol) of phthalimide (1) was added to 4.40 g (22 mmol) of 1,3-dibromoporpane and 6.08 g (44 mmol) of potassium carbonate in 100 mL of DMF. The mixture was stirred at RT for 3 h and then poured into ice. The resulting mixture was filtered and then crystallized from ethanol. Yield 80 %, melting Point 72-74 ºC [12]
2-(3-(4-Phenylpiperazin-1-yl)propyl)isoindoline-1,3-dione (3): 3.00 g (19 mmol) of 1-phenylpiperazine was added to 4.90 g (19 mmol) of 2-(3-bromopropyl)isoindoline-1,3-dione and 6.08 g (44 mmol) of potassium carbonate in 100 mL of acetonitrile. The mixture was allowed to reflux for 3 h. On cooling the acetonitrile was evaporated off and the residue extracted with 150 mL water and 150 mL ethylacetate. The aqueous layer was extracted with 2 × 100 mL ethyl acetate and combined to the organic layer. The solvent was evaporated off to give a pale yellow product. The product was recrystallized from hexane as pale yellow crystals, 4.2 g (12.03 mmol). Yield 65 %, Melting Point 135-137 ºC. ¹H NMR δ (ppm) (CDCl3): 7.87-7.68 (m, 4 H), 7.45-6.81 (m, 5 H), 3.80 (2 H, t, J = 6.90 Hz), 3.13-3.08 (2 H, m), 2.55-2.44 (8 H, m), 1.88 (q, 2 H). MS (ESI) (m/z): [M+H]+ 350.28.
Crystal data and structure determination
A yellow plate single crystal with approximate dimension of 0.37 mm × 0.25 mm × 0.24 mm was mounted on a glass fiber in a random orientation. The data were collected by Bruker APEX-II D8 Venture diffractometer with graphite monochromated Mo Kα radiation (λ =0.071073 nm) using φ and ω scans mode in the range of 2.6° ≤ θ ≤ 30.6° at 293(2) K. A total of 70846 reflections were collected with 5701 unique ones (Rint= 0.055), of which 3507 reflections with [I > 2σ(I)] were considered to be observed and used in the succeeding refinements. The structure was solved by direct methods and expanded by using Fourier differential techniques with SHELXS-97. All non-hydrogen atoms were located with successive difference Fourier syntheses. The structure was refined by full-matrix least-squares method on F2 with anisotropic thermal parameters for all non-hydrogen atoms. Full matrix least-squares refinement gave the final R=0.055 and wR=0.158. Crystal data and structure refinement for the title compound are shown in .
Computational studies
All the quantum chemical calculations in this study were done by applying DFT method with a hybrid functional B3LYP [13] at 6‒31G (d,p) double-zeta basis set with polarization functions on heavy and hydrogen atoms [14] using Gaussian 09 software [15]. The geometry was optimized by energy minimization with respect to all the geometrical parameters without considering any molecular symmetry constraints. Drawing the structure of the optimized geometry and visualization of the HOMO and LUMO calculations have been done by GaussView 5.0.8 program [16].
Evaluation of antimicrobial activity
Antimicrobial activities of the compound 3 were determined by using agar well diffusion method [17]. This was used to examine the susceptibility of the reference microorganisms to the tested compound. Two types of petri dishes were prepared; one with a base layer of Mueller-Hinton agar medium (MHA, Becton Dickinson) and the other with Sabouraud agar. The reference microorganism colonies were suspended in 0.85 % saline solution and the turbidity compared with the 0.5 McFarland standards, to produce a suspension of 1.5 x 108 CFU/mL. Subsequently, equidistant (0.6 cm diameter) holes were made using sterile cork borer in the agar. 100 𝜇L of the tested compound solution at concentration (100 𝜇mol dissolved in 1mL DMSO) was poured in the holes. Ciprofloxacin (50 𝜇g/mL) was used as standard for antibacterial and fluconazole (25 𝜇g/mL) was used as standard for antifungal activity, as positive controls. DMSO solvent was used as negative control. The prepared plates were incubated at 37 oC for 24 h and 48 h for bacterial strains and yeast, respectively. The antibacterial activity of the standard drugs and tested compound was expressed as the mean of inhibition diameters produced in mm. In this study, five reference strains were used: Escherichia coli ATCC 10536, Pseudomonas aeruginosa ATCC 15442, Staphylococcus aureus ATCC 6538, Klebsiella pneumoniae ATCC 13883 and Candida albicans ATCC 90029.
Molecular docking studies
Docking studies was performed for the tested compound in order to understand the mechanism of action as anti-microbial and anti-fungal, molecular modelling and docking studies were performed on X-ray crystal structure of E. coli 24kda domain in complex with clorobiocin (PDB code: 1KZN) [18] and cytochrome P450 14𝛼-sterol demethylase from Mycobacterium tuberculosis (Mycobacterium P450 DM) and fluconazole co-crystalline (PDB code: 1EA1) [19] using Molegro Virtual Docker (MVD 2013.6.0.0 [win32]) program [20].
Results
Crystal structure
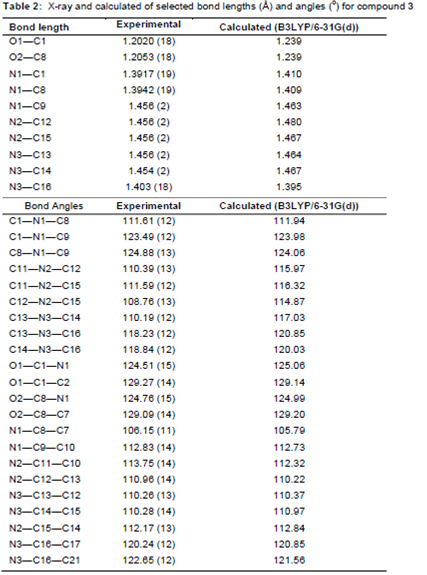
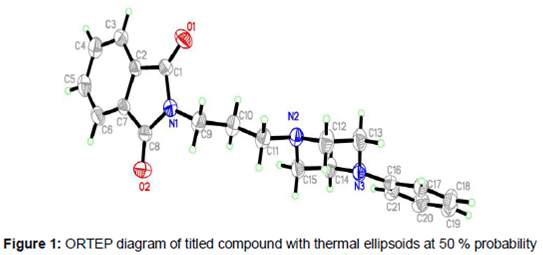
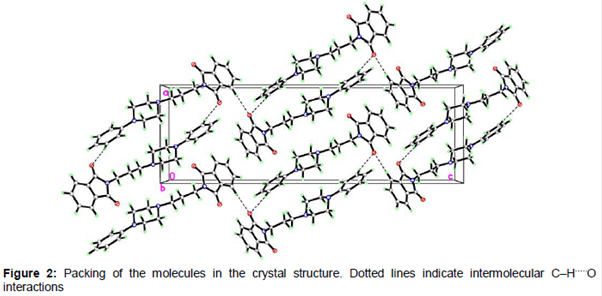
All bond lengths and angles of the compound 3 structure are within the normal ranges (). As shown in , there are two planes in title molecule which are relevant: isoindol ring C1–C8/N1 form plane I and phenyl ring C16-C21 form plane II. The angle between plane I and II is is 79.47 (3)° The isoindol ring system is planar and piperazine ring (N2/C12/C13/N3/C14/C15) adopts an almost perfect chair conformation. The partial crystal packing diagram of molecule revealed the presence of an intermolecular H-bonding network involving C–H and O functionality with neighboring molecules (). The distance of the interactions between H6A···O2 is 2.46 (14) Å and between H20A···O2 is 2.52 (9) Å and the angles C6—H6A···O2 is 169.00 (2)° and C20—H20A···O2 is 152.00 (5).
Total energies, frontier molecular orbitals (FMOs) and chemical reactivity
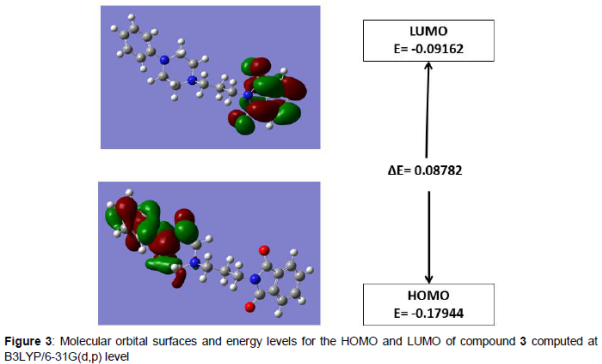
The HOMO and LUMO plots of the title compound are shown in . HOMO of the title compound presents a charge density localized mainly on the benzene rings whereas LUMO is centered upon the O−C−N−C−O of the phthalimide moiety. The calculated energy values of HOMO and LUMO are -0.17944 eV and -0.09162 eV, respectively. The HOMO-LUMO energy gap value is 0.08782 eV.
Antibacterial and antifungal activities
The investigation was carried out three times and the average zone of inhibition was calculated. Compound 3 exhibited a good activity against S. aureus and C. albicans with zones of inhibition of 15 cm and 18 cm, respectively. The remaining strains gave no significant activity with the tested compound.
Molecular docking
In E. coli 24 kda domain, clorobiocin (reference compound) was found to have hydrogen bonding interactions with Asn46 (2.76 Å), and Arg136 (2.96, 3.06, 3.44 Å) with MolDock score -175.0 [22]. The compound 3 revealed a MolDock score of -144.29 and form four hydrogen bonding interactions with Asn46 (3.07, 3.39, 3.46 Å) and Gly119 (3.13 Å) (a). b shows that compound 3 was superimposed with co-crystalline clorobiocin in the active site of the E. coli 24 kDa domain.
Regarding the cytochrome P450 14𝛼-sterol demethylase, the compound 3 revealed a MolDock score -139.15 and form two hydrogen bonding interaction with Arg96 (2.73 Å) and Try76 (2.93 Å)(a). b shows the compound 3 was superimposed with co-crystalline fluconazole in the active site of cytochrome P450 14α-sterol demethylase and this indicate the good quality of the docking study.
Discussion
Most of the optimized bond lengths are slightly higher than the experimental values, due to the molecular states are diverse during the experimental and computed experiments. The theoretical data are calculated in gas phase without molecular interactions are considered, but the experimental data are collected in the solid state and crystal Interactions, such as Van der Waals forces, hydrogen bond interactions and crystal packing force make slight changes in the structure. Anyway, the computed bond lengths and bond angles are in great concurrence with the experimental data. Good correlations were obtained between the calculated and experimental bond lengths and angles 0.994 and 0.942, respectively.
HOMO and LUMO are additionally essential in deciding properties, for example, molecular reactivity and the capacity of a molecule to absorb light. To clarify several types of reaction and for foreseeing the most reactive position in conjugated systems, molecular orbitals and their properties like energy are utilized [23]. LUMO (lowest unoccupied molecular orbital) is the molecular orbital of the lowest energy that is not occupied by electrons and speaks the capacity to obtain an electron; HOMO (highest occupied molecular orbital) is molecular orbital of the highest energy that is possessed by electrons and represents the ability to give an electron. The energy gap of HOMO-LUMO clarify the charge exchange inside the molecule and reflect the biological activity of the molecule [24].
The HOMO-LUMO energy gap value is 0.08782 eV. The small energy gaps are generally connected with a high chemical reactivity, low kinetic stability and these molecules are termed as delicate molecules [25].
Compound 3 gives moderate antibacterial and antifungal activities relative to standard drugs, the experimental results have a good agreement with docking results as the compound 3 give MolDock score smaller than the reference drug co-crystallized in the active site of the target enzymes.
Conclusion
The title compound, 2-(3-(4-phenylpiperazin-1-yl)propyl)isoindoline-1,3-dione (3) has been synthesized successfully and the optimized molecular structure investigated by B3LYP/6-31G (d,p) method using DFT. HOMO–LUMO energy gap of the molecule is 0.08782 eV. Antimicrobial data show moderate activity against S. aureus and C. albicans.
Declarations
Acknowledgement
References
Archives


News Updates